
High-Performance Computing
High-Performance Computing (HPC) provides storage & computational power to support analysis of genomic, transcriptomic, & proteomic data.
Christened Zuputo (meaning ‘The Brain’), the WACCBIP HPC Core is a Dell EMC High Performance Computing Cluster powered by Intel processors with 6 compute nodes comprising 216 CPU cores & 12 NVIDIA Tesla P100 GPU cards. The cluster runs on Mellanox EDR Infiniband interconnect technology and is managed by the ROCKS cluster distribution software.
About Zuputo
System Name | Zuputo |
Nodes | 6 |
Processor | 2x Intel ® Xeon ® Broadwell @ 2.4GHz |
CPU Cores | 216 |
RAM | 1536TB |
GPUs | 12 X NVIDIA TESLA P100 16GB |
Interconnect | Mellanox EDR Infiniband |
Storage | 274 TB Lusture |
Cluster Manager | ROCKS cluster distribution software |
Launch Date | June 22, 2018 |
- Authorisation for access by an organisation
- Authorisation for access by individuals within that organisation
For WACCBIP to grant access to the HPC facility, you are requested to kindly follow the instructions below:
- Read the WACCBIP HPC Terms and Conditions of Use
- Download and fill all details on the form.
- Once completed, sign and email the application form to hpc@ug.edu.gh
Once received, your application will be reviewed, and a decision will be made to either grant access or not as appropriate. You will be notified of the decision.
We look forward to working with you.
The use of High-Performance Computing (HPC) resources is subject to the IT Policies of University of Ghana Computing Systems (UGCS). In addition, the following policies shall also govern the use of the HPC facilities to avoid misuse of computing resources. Applicants are required to agree to follow these guidelines before they are given a user account that will grant them access to the system. Legal or administrative action(s) shall be taken against any user who violates the rules.
HPC Account Policy
- The HPC infrastructure is meant to be accessible only to registered users who have agreed to the stipulated terms and conditions.
- All users shall be responsible for protecting their accounts from unauthorized access.
- Users are not allowed to share their credentials with others under any condition, including via .rhost
- Users are prohibited from using the credentials of others to access the HPC system.
- The University of Ghana reserves the right to
- Create an HPC account
- Deactivate/Close or Suspend an HPC account if it is determined to have used HPC resources for purposes other than those allowed. .
- Modify an account
- University of Ghana Reserves the right to accept or reject any request for an HPC account.
HPC Resource Usage Policy
- HPC resources must be used for academic and research purposes only.
- The HPC system should not be used for activities that lead to personal or private (commercial) benefit.
- Use of the HPC system for activities that violate the laws of Ghana is prohibited.
- Resources must not be used to support illegal or malicious activities including but not limited to: A) Production of biological, nuclear or chemical weapons or any activity capable of producing any other types of weapons.B) Acts of terrorism.
- Users are only permitted to access the login node. This should be done via a secure communication channel such as Secure Shell(ssh). Access is prohibited to other nodes such as the compute nodes, master nodes, storage nodes and management nodes.
- All jobs and processing shall occur in the login node. The login node shall be used for interactive work. Heavy computational work should be submitted to the compute nodes via a Job Scheduler(OpenPBS). Users shall not under any condition access the compute nodes directly.
- The use of the HPC resources to access, store, manipulate unauthorized data or programs remotely or on the HPC system is prohibited.
- Users shall not under any circumstance access, store, upload, download, transmit or create sexually explicit or pornographic material on/from/to the HPC system.
- This account policy is subject to renewal and amendment.
Maintenance Policy
- UGCS shall inform users of HPC system maintenance via email messages. However, UGCS reserves the right to shut down the HPC system without prior notice in case of maintenance or emergency.
Security Policy
- Users are obliged to report any computer security flaws or weaknesses, incidents of misuse or violation of the HPC user account policies to the System Administrator.
- Users shall not under any circumstance perform any activity that will lead to circumvention of security or administrative measures and policies.
- Users shall not purposely make copies of system or configuration files (such as password file, MOM files, ) for any unauthorized activity.
- Download or Execution of programs which reveal security flaws of the HPC system is prohibited.
Monitoring and Privacy
- University of Ghana reserves the right to perform the following activities without a user’s prior notice, knowledge or consent:i) Monitor all accounts/activities of users/resources/data//files of the HPC system and networks at any time ii) Retain copies of data/messages/other files stored/accessed/transferred/modified in or transferred from the HPC system iii) Share all information gathered with Government or Law enforcement agencies
- Users will have no explicit or implicit privacy while using their user account.
Data Usage Policy
- Modification of data on the HPC system without authorization is prohibited.
- Users shall not upload or store data which violates any copyright agreements/laws.
- University of Ghana reserves the right to remove or stop the transfer of any data at any time after a user account has expired, been deleted or no longer has a research association with the University.
- Users are responsible for keeping their data intact.
Data Retention Policy
- UG reserves the right to make copies of any data for backup purposes/retain such data indefinitely/stop backup of data/delete data.
User Data / Account
- HPC user accounts will be deactivated if they are inactive (no logins) for 6 months.Files associated with the deactivated account (home/username) will be retained for 3 months after deactivation. During the 3 months, users can request for a reactivation of their account.
- Files in temp_dirare deleted after 28 days (from date they were last accessed)
- User accounts that are associated with projects will be deactivated once the project expires. However, the data will be retained for a maximum period of 12 months after which UG reserves the right to reclaim storage space by permanently deleting the data.
- Users have the option to arrange for a permanent data storage by contacting the System Administrator.
User Abuse Policy
- An abusive user is defined by UGCS as a user who makes it difficult for others to use the system by:A) Circumventing controls like job scheduling policy B) Making shared files accessible to others C) Manipulating the system to their benefit.
If a user is found to be abusing the system the following sanctions shall be applied:
First Incident
- A notification to desist will be sent to the user by e-mail, phone or text message.
- A user’s job may be terminated, if necessary, to return the system to normal.
- A user’s ability to submit/run jobs or store data will be suspended.
- A user’s ability to submit/run jobs or store data will be returned, once the user stops the act(s).
Second incident
- If a user should repeat the abusive act(s), his superior/manager/supervisor shall be informed of the act.
- All other measures in first incident will be applied.
Third incident
- User’s account shall be locked for 3 – 12 months, after which UGCS shall decide whether to enable the account or disable the account permanently.
Acknowledgement
Any work that involved the use of Zuputo should be referenced as
- Conference name
- Title of talk/poster/presentation/book/article
- Author(s)
- Journal, volume,
- Pages or editors
- Address
- Publisher
- Year
- DOI
All jobs are run using PBS Scheduler. This is a software that enables the user to select resources needed for the task.
Resources that you can request for are : Nodes, Processors, GPUs, RAM, Walltime.
Jobs can be run using any of the following:
An interactive PBS job can be requested for any activity which needs an environment the same as for a batch job. After a successful request, you will get a prompt, and access to all the resources assigned to the job. This is very handy when testing or debugging . For example running MPI jobs that across multiple nodes.
There are two ways you can submit an interactive job to PBS: By including all of the options on the command line, or by listing the options in a PBS script and submitting the script with the command-line option specifying an interactive job.
Submitting an interactive job from the command line
To submit from the command line, you need to specify all of the PBS options that would normally be specified by directives in a PBS script(Exclude #PBS ). Use the qsub command with -I argument
$ qsub -I
For interactive jobs you can still additional options such as the resource options for number of nodes, processors, memory, and walltime, and the -V option to insure that all the nodes get the correct environment. The -I flag signals that the job should run as an interactive job. (Note: in the example that follows, the character indicates that the following line is a continuation of the one on which it appears.)
$ qsub -I -V -q workq -l nodes=2:ppn=2 -l mem=1GB -l walltime=2:00:00
The above example requests an interactive job in the workq queue with the following resources: two nodes with two processors, each processor with 1 GB of memory for two hours. The prompt will change to something that says the job is waiting to start, followed by a prompt on the first of the assigned nodes. You can always omit the -q option because the system automatically allocates the job to a queue
qsub: waiting for job 12345678.master1.local to start qsub job 12345678.master1.local ready To add GPUs, add the ngpus argument $ qsub -I -V -q flux -l nodes=2:ppn=2 -l mem=1GB -l walltime=4:00:00 -l ngpus=2
If at some point before the interactive job has started you decide you do not want to use it, the command Ctrl-C will cancel it
When you have completed the work for which you requested the interactive job, you can just logout of the compute node, either with exit or with logout, and you will return to the login node prompt.
[user@login ~]$ exit [user@login ~]$ logout
Submitting an interactive job using a file
To recreate the same interactive job as above, you could create a file, say job.pbs, with the following lines in it
#!/bin/bash #PBS -l nodes=2:ppn=2 #PBS -l mem=1GB #PBS -l walltime=2:00:00 #PBS -l ngpus=2
then submit the job using
$ qsub -I job.pbs
To run a batch job on the HPC cluster, you will need to set up a job submission script. This is a Portable Batch System (PBS) file. This PBS is a simple text file that defines the commands and cluster resources used for the job. A PBS file can be easily created or edited with a UNIX editor, such as emacs,pico or vi, pico.Should in case you write your script in windows environment and you upload to the server. Format the file with the dos2unix utility.
For example, if you newly created file is job.pbs , then you format with the command
dos2unix job.pbs
Submitting a Job
In order to use the HPC compute nodes, you must first log in to the server, and submit a PBS job. Do not run jobs on the login node. The qsub command is used to submit a job to the PBS queue, delete an already submitted job and to request additional resources. The qstat command is used to check on the status of a job already in the PBS queue. To simplify submitting a job, you can create a PBS script and use the qsub and qstat commands to interact with the PBS queue.
Creating a PBS Script
To set the parameters for your job, you can create a control file that contains the commands to be executed. Typically, this is in the form of a PBS script. This script is then submitted to PBS using the qsub command.
Here is a sample PBS file, named myjobs.pbs, followed by an explanation of each line of the file.
#!/bin/bash #PBS -l nodes=1:ppn=2 #PBS -l mem=1GB #PBS -l ngpus=2 #PBS -l walltime=00:00:45 cd /home/waccbip/test/ echo 'hello world'>myfile.txt
- The first line in the file identifies which shell will be used for the job. In this example, bash is used but csh or other valid shells would also work.
- The second line specifies the number of nodes and processors desired for this job. In this example, one node with two processors is being requested.
- The third line specifies the RAM the use r requires for the job. In this example a 1GB RAM has been requested
- The fourth line states how many GPUs is needed . In this example 2GPUs are requested.
- The fifth line in the PBS file states how much wall-clock time is being requested. The format is hh:mm:ss. In this example 45 seconds of wall time have been requested.
- The sixth line tells the HPC cluster to access the directory where the data is located for this job. In this example, the cluster is instructed to change the directory to the /home/waccbip/test/ directory.
- The fifth line saves the text ‘hello world’ to a text file called myfile.txt
Submitting the job using the qsub command
To submit your job without requesting additional resources, issue the command
$qsub myjob.pbs
Requesting for additional wall time
If you need additional or less wall time for job execution for a submitted job, use the qsub command to make the request
The example script above, requested for 45 seconds.If you realize that you need more time say,3 minutes then you can make the changes using this command:
$qsub -l walltime=0:03:00 myjob.pbs
3 minutes will be requested from PBS. Note: If a job does not finish within the specified time, it will be terminated.
Requesting Nodes and Processors
Sometimes you may wish to alter the number of nodes and processors requested for a job. This can also be done by using the qsub command. In the example script, we have requested one node with two processors, or one dual-processor node.
If you realize later that you need three HPC nodes for your job but you are going to use only one of the dual-processors on each node, then use the following command:
$qsub -l walltime=0:03:00,nodes=3 myjob.pbs
If you want to use both processors on each HPC node, you should use the following command:
$qsub -l walltime=0:05:00,nodes=3:ppn=2 myjob.pbs
Requesting GPU nodes
You can submit jobs directly to the graphical processing unit (GPU) cluster by specifying the number of GPUs your job requires in your PBS request.
For example, if you need four nodes with 1 cores per node and two GPUs per node, then your command should look like this:
$qsub -l walltime=0:03:00,nodes=4:ppn=16 -l ngpus=2
Several software packages are installed on the clusters. These softwares can be accessed and used using module files. Modules allow you to specify which versions of the packages you want.
To list all available module files use the command below
module avail
You can also list versions available for a particular software or all module files whose name contains a specified string For example, to find all all versions available for python, use:
module avail python/
To use a module :
module load modulename
To load Python version 3.5.5 use this command:
module load python/3.5.5
(replace modulename with the name of your module of interest)
You can also load multiple loads with a singe command . eg.
module load samtools bwa python
To unload the module execute the command:
module unload modulename
Example, to unload python version 3.5.5
module unload python/3.5.5
If you want to unload all softwares in your environment execute the command:
module purge
List all loaded module files by executing the command:
module list
If you don’t find your required software in the list of modules, you can use anaconda to install the package or send a request to hpc@ug.edu.gh
Getting more information
If you want to get a brief description of a module use the following commands:
module whatis modulename
module help modulename
Module collections
Sometimes you might want to run two or more sets of modules separately due conflicts. You might also have a long list of modules to load every time you log on to the cluster.
In such situations module collections can be used. Module collections allow you to create saved environments that remember a set of modules to load together.
First, load all your modules and then create the saved environment using the command:
module save
The set of modules will be saved as your default set. If you want to save a set as a non-default, assign the environment a name:
module save environment-name
To load all the modules in the set(your default), use the command:
module restore
To load a non-default list, just add the environment name
module restore environment-name
Get a list of your saved environments using:
module savelist
System wide software packages are available via the environmental modules. However, you might not get all the software you need. To install additional software packages use the conda environment.
Conda is a package manager that makes software installation easy. It can be used to install standalone softwares such as bwa, gromacs, R. It is also possible to install packages which are dependent on other softwares. For example numpy and tensorflow which work with python can be installed using conda( but of course python the dependency python must first be installed)
In conda, software packages are installed via channels which are software repositories .An example of such a channel is Bioconda which contains over 3000 bioinformatics software. The list of software is available can be found here
1. Load anaconda
module load anaconda/3
2. Setting up channels
Conda comes with a default channel. But this might not contain the softwares you need. Other popular channels are bioconda and conda-forge. Add channels using the command:
conda config --add channels channel-name
Lets add the channels bioconda and conda-forge
conda config --add channels bioconda
conda config --add channels conda-forge
To view channels, use the command:
conda config --get channels
Read more about setting channels here
To remove a channel lets say conda-forge Use the command below
conda config --remove channels conda-forge
3. Install packages
Once your channels have been enabled, you can install any package available on the channels into the current conda environment.
conda install samtools
If you experience any issue(usually permission issues), then you need to activate an environment before installing the software.
source activate environment conda install samtools
You can also create a new environment and install the software package.
conda create -n alignment muscle
To install a package from a specific channel use:
conda install -c channel-name package-name
Example is the installation of gromacs from bioconda channel
conda install -c bioconda gromacs
List all install packages using the command
conda list
To list all environments use:
conda env list
Resources
To transfer files to and from Zuputo, the scp command is used. The format is
scp -P <port number> <source> <destination>
Lets look at some examples
Transfering files from Zuputo to a local machine
if path to the file on the server is myhome/file
then to transfer the to the local machine
scp -P <port number> <username@zuputo.ug.edu.gh:myhome/file <destination on local machine>
to transfer the entire contents of a directory to your local machine
scp -P <port number> -r <username@zuputo.ug.edu.gh:myhome/file <destination on local machine>
Transfering files from local machine to Zuputo
The same procedure as above but this time the destination becomes <username@zuputo.ug.edu.gh:myhome/directory>
Step 1: Preparing the conda environment
Load anaconda
module load anaconda
, and check which python executable is used, e.g.
which python /opt/apps/anaconda3/bin/python
Anaconda provides ready-to-use python packages but it also useful to use the conda environment because
1. It creates seperate environments in which all package dependencies can be installed without affecting other environment configurations;
2. It allows users to install packages without administrative permission.
After the anaconda module is loaded, create your environment . When creating the environment you can add some packages to be installed.
conda create --name myenv pip numpy
The above command creates an environment called myenv whiles installing pip and numpy at the same time
To use the environment , you need to activate with the command
source activate myenv
To deactivate the environment use
source deactivate myenv
Note: Before you do all these activities you should load anaconda
Step 2: Installing Python Packages
After creating the environment, python packages are installed using pip( other methods are available)
Activate your environment
Install packages using pip
pip install <package>
It is possible to install two or more packages in single command
pip install <package1> <package2> <package3>
Note
The created conda environment is saved in your home directory under the path $HOME/.conda/envs. Environments are organised in different subfolders so each time a new package is installed , the relevant files will also be created in its own subfolder. The conda environments takes space from the quota of your home directory.
You can check where the packages are using the command
which <package>
Step 3: Submitting your job
You first need to create your python script
Then create your job submission script to run the python script
Introduction
Genome annotation is done on a draft genome assembly. The main aim of genome annotation is to add biological information to the newly assembled genome which is “blank” so far as information is concerned.
For a bacterial genome we will be interested in the following information (which is non-exhaustive at this point):
– protein coding genes i.e promoter regions, robosome binding sites,coding sequence ,etc
-non coding genes (such as tRNA,rRNA, etc)
How do we perform Annotation?
There are a number of tools and pipelines that have been developed specifically for this purpose (including RAST and Prokka). In this tutorial we look at the use of prokka for annotation. Now lets dig in
- Prepare your job submission script
#!/bin/bash
#PBS –l nodes=1:ppn=10
#PBS –l mem=32GB
- Load prokka into your environment
module load bioinfo/prokka/1.12
- Download fasta file
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/013/925/GCA_000013925.2_ASM1392v2/GCA_000013925.2_ASM1392v2_genomic.fna.gz
- Unzip it
gunzip GCA_000013925.2_ASM1392v2_genomic.fna.gz
- Annotate with prokka
prokka --cpus 10 --kingdom Bacteria --prefix Agy99 --genus Mycobacterium --locustag GCA_000013925 GCA_000013925.2_ASM1392v2_genomic.fna
- There should be a folder called Agy99 created. cd to it
cd Agy99
In the folder 12 files will be created.
Agy99.err, Agy99.faa, Agy99.ffn, Agy99.fna, Agy99.fsa, Agy99.gbk, Agy99.gff, Agy99.log, Agy99.sqn, Agy99.tbl, Agy99.tsv, Agy99.txt
Now we can search and count some genes
Count the number of annotations created
grep ‘>’ Agy99.ffn | wc –l
Count the number of tRNA
grep ‘tRNA’ Agy99.ffn | wc –l
Count the number of hypothetical proteins
grep 'hypothetical protein’ Agy99.faa | wc –l
Congratulations. You have successfully performed a genome annotation. CHEERS!!!!!!!!!!
Installation
There is a system wide installation which can be loaded via the module file
User specific installation must be done via anaconda package
conda install –c conda-forge –c bioconda –c defaults prokka
(Note: This must be done after activating your conda environment)
RNA-SEQ analysis enables us to identify genes or transcripts that are differentially expressed across samples. It also makes it possible to identify novel transcripts and multiple isoforms for gene. This tutorial provides a step by step instructions on how to perform such analysis.
Step 1: prepare your analysis environment .
Load anaconda package
module load anaconda/3
Add channels
conda config --add channels conda-forge conda config --add channels defaults conda config --add channels r conda config --add channels bioconda
create a new environment
conda create -n rna-seq r-essentials r-base python=3.7
Activate the environment
source activate rna-seq
Install the packages
conda install -c bioconda hisat2 conda install -c bioconda samtools conda install -c bioconda stringtie conda install -c bioconda gffcompare conda install -c bioconda bioconductor-biocinstaller conda install -c conda-forge r-Kindly issue the other gert conda install -c conda-forge r-devtools / conda install -c r r-devtools conda install -c r r-dplyr
Open R terminal
R
Install R packages
devtools::install_github("alyssafrazee/RSkittleBrewer")
install.packages(‘BiocManager’)
BiocManager::install(‘ballgown’)
BiocManager::install('genefilter')
BiocManager::install(‘cowplot’)
Step 2: Data acquisition
Download the data available from here. (data size is about 2GB)
wget ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/chrX_data.tar.gz
Unpack the data (Data will be stored in chrX_data)
tar xvfz chrX_data.tar.gz
Data will be extracted to a folder called chrX_data which will have the structure samples/indexes/genome/genes . You can check this, using the command:
tree chrX_dataData description
- A a paired-end end sequence of the human chromosome X located in chrX_data/samples/*.fastq.gz. This will be our raw reads.
- A reference genome (chromosome X) located in chrX_data/genome.chrX.fa.
- A description of the data is stored a file geuvadis_phenodata.csv. We will refer to this is the phenotypic data. This will be used in the differential analysis step and is usually created by yourself using your samples.
- Reference Genome annotations located chrX_data/genes/chrX.gtf
- Index files. We will however generate our own index files.
Step 3: Start Analysis
Map reads to reference genome. This is done to assign reads to specific regions in the genome. Prior to mapping we will have to first index the reference genome
Create a folder index
mkdir index
Extract exon and splice-site information from the genome annotation
extract_splice_sites.py chrX_data/genes/chrX.gtf > index/chrX.ss extract_exons.py chrX_data/genes/chrX.gtf > index/chrX.exon
Confirm that these files have been created
head -n 5 index/chrX.ss
chrX 276393 281481 +
chrX 281683 284166 +
chrX 284313 288732 +
chrX 288868 290647 +
chrX 290775 291498 +
head -n 5 index/chrX.exon
chrX 276323 276393 +
chrX 281393 281683 +
chrX 284166 284313 +
chrX 288732 288868 +
chrX 290647 290775 +
Build index using hisat2. Syntax for building index using hisat2
histat2-build -p <number_of_threads> --ss <split_sites> --exon <exon> <reference.fa> <index_basename>
Note: –ss and –exon flags can be omitted if gene annotations is unavailable.
Now its time to build the index
hisat2-build -p 10 --ss index/chrX.ss --exon index/chrX.exon chrX_data/genome/chrX.fa index/chrX_index
Confirm that the index files have been created
ls -1 index
chrX.1.ht2
chrX.2.ht2
chrX.3.ht2
chrX.4.ht2
chrX.5.ht2
chrX.6.ht2
chrX.7.ht2
chrX.8.ht2
chrX.exon
chrX.ss
Before we do the mapping let's find out how many samples we are dealing with.
ls chrX_data/samples/*chrX_1*.gz |wc -l
12
Syntax for hisat2 mapping
hisat2 -p <number_of_threads> --dta -x <index_basename> -1 <Read1> -2 <Read2> -S <outputname_for_generated_samfile>
We create a folder where all the generated samfiles will be stored
mkdir map
Now let's do the mapping for all the samples
hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188044_chrX_1.fastq.gz -2 chrX_data/samples/ERR188044_chrX_2.fastq.gz -S map/ERR188044_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188104_chrX_1.fastq.gz -2 chrX_data/samples/ERR188104_chrX_2.fastq.gz -S map/ERR188104_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188234_chrX_1.fastq.gz -2 chrX_data/samples/ERR188234_chrX_2.fastq.gz -S map/ERR188234_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188245_chrX_1.fastq.gz -2 chrX_data/samples/ERR188245_chrX_2.fastq.gz -S map/ERR188245_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188257_chrX_1.fastq.gz -2 chrX_data/samples/ERR188257_chrX_2.fastq.gz -S map/ERR188257_chrX.sam
hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188273_chrX_1.fastq.gz -2 chrX_data/samples/ERR188273_chrX_2.fastq.gz -S map/ERR188273_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188337_chrX_1.fastq.gz -2 chrX_data/samples/ERR188337_chrX_2.fastq.gz -S map/ERR188337_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188383_chrX_1.fastq.gz -2 chrX_data/samples/ERR188383_chrX_2.fastq.gz -S map/ERR188383_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188401_chrX_1.fastq.gz -2 chrX_data/samples/ERR188401_chrX_2.fastq.gz -S map/ERR188401_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188428_chrX_1.fastq.gz -2 chrX_data/samples/ERR188428_chrX_2.fastq.gz -S map/ERR188428_chrX.sam hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188454_chrX_1.fastq.gz -2 chrX_data/samples/ERR188454_chrX_2.fastq.gz -S map/ERR188454_chrX.sam
hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR204916_chrX_1.fastq.gz -2 chrX_data/samples/ERR204916_chrX_2.fastq.gz -S map/ERR204916_chrX.sam
Convert the SAM files to sorted bam files using samtools. This is done because bam files are usually compressed and more efficient to work with than SAM files
samtools sort -@ 8 -o map/ERR188044_chrX.bam map/ERR188044_chrX.sam samtools sort -@ 8 -o map/ERR188104_chrX.bam map/ERR188104_chrX.sam samtools sort -@ 8 -o map/ERR188234_chrX.bam map/ERR188234_chrX.sam samtools sort -@ 8 -o map/ERR188245_chrX.bam map/ERR188245_chrX.sam samtools sort -@ 8 -o map/ERR188257_chrX.bam map/ERR188257_chrX.sam samtools sort -@ 8 -o map/ERR188273_chrX.bam map/ERR188273_chrX.sam samtools sort -@ 8 -o map/ERR188337_chrX.bam map/ERR188337_chrX.sam samtools sort -@ 8 -o map/ERR188383_chrX.bam map/ERR188383_chrX.sam samtools sort -@ 8 -o map/ERR188401_chrX.bam map/ERR188401_chrX.sam samtools sort -@ 8 -o map/ERR188428_chrX.bam map/ERR188428_chrX.sam samtools sort -@ 8 -o map/ERR188454_chrX.bam map/ERR188454_chrX.sam samtools sort -@ 8 -o map/ERR204916_chrX.bam map/ERR204916_chrX.sam
Let's delete the SAM files to clear up storage space.
rm map/*.sam
Assemble transcripts using stringtie
# create assembly per sample using 8 threads
stringtie map/ERR188044_chrX.bam -l ERR188044 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188044_chrX.gtf stringtie map/ERR188104_chrX.bam -l ERR188104 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188104_chrX.gtf stringtie map/ERR188234_chrX.bam -l ERR188234 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188234_chrX.gtf stringtie map/ERR188245_chrX.bam -l ERR188245 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188245_chrX.gtf stringtie map/ERR188257_chrX.bam -l ERR188257 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188257_chrX.gtf stringtie map/ERR188273_chrX.bam -l ERR188273 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188273_chrX.gtf stringtie map/ERR188337_chrX.bam -l ERR188337 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188337_chrX.gtf stringtie map/ERR188383_chrX.bam -l ERR188383 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188383_chrX.gtf stringtie map/ERR188401_chrX.bam -l ERR188401 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188401_chrX.gtf stringtie map/ERR188428_chrX.bam -l ERR188428 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188428_chrX.gtf stringtie map/ERR188454_chrX.bam -l ERR188454 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR188454_chrX.gtf stringtie map/ERR204916_chrX.bam -l ERR204916 -p 8 -G chrX_data/genes/chrX.gtf -o assembly/ERR204916_chrX.gtf
Merge transcripts from all samples
We first put the names of the assembled transcript files into a single file
ls assembly/*.gtf > mergelist.txt
cat mergelist.txt
assembly/ERR188044_chrX.gtf assembly/ERR188104_chrX.gtf assembly/ERR188234_chrX.gtf assembly/ERR188245_chrX.gtf assembly/ERR188257_chrX.gtf assembly/ERR188273_chrX.gtf assembly/ERR188337_chrX.gtf assembly/ERR188383_chrX.gtf assembly/ERR188401_chrX.gtf assembly/ERR188428_chrX.gtf assembly/ERR188454_chrX.gtf assembly/ERR204916_chrX.gtf
Syntax for merging stringtie -p <number_of_threads> -G <genome_annotation> -o <outputfile> <textile_containing_list_of_transcripts>
We can now merge with stringtie
stringtie --merge -p 8 -G chrX_data/genes/chrX.gtf -o stringtie_merged.gtf mergelist.txt
Let's confirm the operation was successful
cat stringtie_merged.gtf | head -n 5
# StringTie version 2.1.4
chrX StringTie transcript 323525 324046 1000 + . gene_id “MSTRG.1”; transcript_id “MSTRG.1.1”;
chrX StringTie exon 323525 323661 1000 + . gene_id “MSTRG.1”; transcript_id “MSTRG.1.1”; exon_number “1”;
chrX StringTie exon 323905 324046 1000 + . gene_id “MSTRG.1”; transcript_id “MSTRG.1.1”; exon_number “2”;
We can find how many transcripts were identified.
cat stringtie_merged.gtf |cut -d$'\t' -f 3|grep 'transcript'|wc -l
4602
We can also inspect how transcripts identified by stringtie compare with what is in the reference annotation using gffcompare tool
gffcompare -r chrX_data/genes/chrX.gtf -G -o merged stringtie_merged.gtf
cat merged.stats
# gffcompare v0.11.5 | Command line was:
#gffcompare -r chrX_data/genes/chrX.gtf -G -o merged stringtie_merged.gtf
#
#= Summary for dataset: stringtie_merged.gtf
# Query mRNAs : 4602 in 1410 loci (4225 multi-exon transcripts)
# (664 multi-transcript loci, ~3.3 transcripts per locus)
# Reference mRNAs : 2102 in 1086 loci (1856 multi-exon)
# Super-loci w/ reference transcripts: 1065
#—————– | Sensitivity | Precision |
Base level: 100.0 | 76.9 |
Exon level: 98.2 | 73.9 |
Intron level: 100.0 | 78.1 |
Intron chain level: 100.0 | 43.9 |
Transcript level: 99.2 | 45.3 |
Locus level: 98.9 | 74.8 |
Matching intron chains: 1856
Matching transcripts: 2085
Matching loci: 1074
Missed exons: 0/8804 ( 0.0%)
Novel exons: 1862/12363 ( 15.1%)
Missed introns: 0/7946 ( 0.0%)
Novel introns: 780/10175 ( 7.7%)
Missed loci: 0/1086 ( 0.0%)
Novel loci: 345/1410 ( 24.5%)\
Total union super-loci across all input datasets: 1410
4602 out of 4602 consensus transcripts written in merged.annotated.gtf (0 discarded as redundant)
The high sensitivity of the transcript level indicates that large percentage of the stringtie transcripts match that of the reference annotation. The lower precision value indicates that many of the transcripts identified by stringtie are not in the reference annotation.This indicates the transcripts are either novel or false positives
The novel exons, introns, and loci indicate how many of the sites were not found in the reference annotation.
Estimate transcript abundance
mkdir abundance stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188044/ERR188044_chrX.gtf map/ERR188044_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188104/ERR188104_chrX.gtf map/ERR188104_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188234/ERR188234_chrX.gtf map/ERR188234_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188245/ERR188245_chrX.gtf map/ERR188245_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188257/ERR188257_chrX.gtf map/ERR188257_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188273/ERR188273_chrX.gtf map/ERR188273_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188337/ERR188337_chrX.gtf map/ERR188337_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188383/ERR188383_chrX.gtf map/ERR188383_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188401/ERR188401_chrX.gtf map/ERR188401_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188428/ERR188428_chrX.gtf map/ERR188428_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR188454/ERR188454_chrX.gtf map/ERR188454_chrX.bam stringtie -e -B -p 8 -G stringtie_merged.gtf -o abundance/ERR204916/ERR204916_chrX.gtf map/ERR204916_chrX.bam
Differential expression
To run the differential expression analysis we use some R packages. First we load our R and call the relevant packages.
R
library(devtools)
library(ballgown)
library(genefilter)
library(dplyr)
library(RSkittleBrewer)
We load the phenotype data
pheno_data <- read.csv(“chrX_data/geuvadis_phenodata.csv”)
#Let's show information for first 6 samples
head(pheno_data)
ids sex population
1 ERR188044 male YRI
2 ERR188104 male YRI
3 ERR188234 female YRI
4 ERR188245 female GBR
5 ERR188257 male GBR
6 ERR188273 female YRI
Load the expression data (i.e. transcript abundance estimates) using the ballgown library. We specify three parameters:dataDir for specify where the estimates are stored, samplePattern to indicate a name pattern that is present in the sample names and pData which specifies the phenotypic information (we loaded it as pheno_data).
bg_chrX <- ballgown(dataDir = 'abundance', samplePattern = 'ERR', pData=pheno_data)
We can check some information about the load abundances
class(bg_chrX)
[1] “ballgown”
attr(,”package”)
[1] “ballgown”
bg_chrX
ballgown instance with 3491 transcripts and 12 samples
We can display expression levels for genes, transcripts,exons and introns using the functions gexpr,texpr, eexpr(), and iexpr() respectively
To get the gene expression levels
head(gexpr(bg_chrX), 3)
FPKM.ERR188044 FPKM.ERR188104 FPKM.ERR188234 FPKM.ERR188245
MSTRG.1 9.153040 0 7.683855 0
MSTRG.10 8.467854 0 0.000000 0
MSTRG.100 0.000000 0 0.000000 0
FPKM.ERR188257 FPKM.ERR188273 FPKM.ERR188337 FPKM.ERR188383
MSTRG.1 0 0 2.417433 0
MSTRG.10 0 0 0.000000 0
MSTRG.100 0 0 0.000000 0
To get transcript expression levels
head(texpr(bg_chrX),3)
FPKM.ERR188044 FPKM.ERR188104 FPKM.ERR188234 FPKM.ERR188245 FPKM.ERR188257
1 8.467854 0 0.00000 0.00000 0.00000
2 0.000000 0 13.99012 14.15741 27.59774
3 0.000000 0 0.00000 0.00000 53.61156
FPKM.ERR188273 FPKM.ERR188337 FPKM.ERR188383 FPKM.ERR188401 FPKM.ERR188428
1 0.00000 0.00000 0.00000 4.871071 0.00
2 0.00000 0.00000 0.00000 21.825052 0.00
3 18.42071 83.10745 27.19952 0.000000 21.08
FPKM.ERR188454 FPKM.ERR204916
1 0.00000 0.00000
2 0.00000 5.52904
3 74.33337 41.66899
We now filter out to remove low abundance genes/transcripts. This is done to remove some genes that have few counts. Filtering out improves the statistical power of differential expression analysis. We use variance filter to remove transcripts with low variance( 1 or less)
bg_chrX_filt=subset(bg_chrX,"rowVars(texpr(bg_chrX)) >1",genomesubset=TRUE)
# 1,264 transcripts were filtered out
Let's check how many transcripts were filtered out. We will check how many trancripts are in the unfiltered and filtered expression data
bg_chrX_filt
ballgown instance with 3250 transcripts and 12 samples
bg_chrX
ballgown instance with 4602 transcripts and 12 sample
If you do the maths , we get 1352 transcript filtered out
Finally, we perform differential expression analysis to reveal transcripts that show significant difference across groups. This is done using the stattest function. The parameters we use are, feature, covariate, adjustvars, getFC and meas. Our feature of interest is transcript we are interested in its differential expressed in males and females so our covariate becomes the sex group. Transcripts might also be differentially expressed among differential populations (confounders) but since this is not our group of interest we have to correct for that using the adjustvars parameter. We also want to get the confounder-adjusted fold change values so we set getFC to TRUE. Finally ballgown gives the expression values in different measures but we will use FPKM as the meas. Testing for differential expression at exons and introns can also be done by changing the feature parameter.
Let's perform the differential analysis on transcripts. The results will be in an data frame
Let's check the first 6 transcripts and their values
head(results_transcripts)
feature id fc pval qval
1 transcript 1 1.7614945 0.2498894 0.8639175
2 transcript 2 0.8250292 0.8471261 0.9783267
3 transcript 3 0.6453535 0.7493203 0.9554001
4 transcript 4 2.7530478 0.1076059 0.8356592
5 transcript 5 1.4828120 0.5873142 0.8934238
6 transcript 6 0.4398648 0.2000345 0.8639175
To check how many of the transcripts have qvalue <0.05 we use the command
table(results_transcripts$qval < 0.05)
FALSE TRUE
3237 13
Let's perform the differentially analysis on genes
results_genes <- stattest(bg_chrX_filt,feature="gene",covariate="sex",adjustvars = c("population"),getFC=TRUE, meas="FPKM")
Let's check the first 6 genes
head(results_genes)
feature id fc pval qval
1 gene MSTRG.1 0.8590737 0.8277795 0.9766457
2 gene MSTRG.10 1.8171603 0.2566553 0.7974964
3 gene MSTRG.100 1.4437594 0.4360308 0.8692654
4 gene MSTRG.1000 1.4264802 0.1599910 0.7207850
5 gene MSTRG.1001 1.1644814 0.4806989 0.8692654
6 gene MSTRG.1002 0.9779751 0.9519060 0.9867803
Find out how many of the genes have a qvalue < 0.05
table(results_genes$qval<0.05)
FALSE TRUE
889 9
The results_transcipts data does not have gene ID so we add that information to it.
results_transcripts = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt), geneIDs=ballgown::geneIDs(bg_chrX_filt), results_transcripts)
Let's confirm that the IDs have been added
head(results_transcripts)
geneNames geneIDs feature id fc pval qval
1 . MSTRG.10 transcript 1 1.7614945 0.2498894 0.8639175
2 PLCXD1 MSTRG.11 transcript 2 0.8250292 0.8471261 0.9783267
3 . MSTRG.11 transcript 3 0.6453535 0.7493203 0.9554001
4 . MSTRG.11 transcript 4 2.7530478 0.1076059 0.8356592
5 . MSTRG.12 transcript 5 1.4828120 0.5873142 0.8934238
6 . MSTRG.12 transcript 6 0.4398648 0.2000345 0.8639175
Let's arrange the results from the smallest P value to the largest
results_transcripts = arrange(results_transcripts,pval)
head(results_transcripts)
results_genes = arrange(results_genes,pval)
head(results_genes)
We can now save the results to a file (csv recommended)
write.csv(results_transcripts, “fc_transcript_results.csv”, row.names=FALSE)
write.csv(results_genes, “fc_gene_results.csv”, row.names=FALSE)
Let's subset transcripts that are detected as differentially expressed at qval <0.05
subset_transcripts <- subset(results_transcripts,results_transcripts$qval<0.05)
subset_genes <- subset(results_genes,results_genes$qval<0.05)
Save the subsets to a file
write.csv(subset_transcripts, “fc_transcript_subset.csv”, row.names=FALSE)
write.csv(subset_genes, “fc_gene_subset.csv”, row.names=FALSE)
Let's save the dge results to a file
write.csv(dge_results,foldchanges.csv,row.names=FALSE)
Now let's confirm that the files have been saved successfully.
#Save to file
write.csv(subset_transcripts, “fc_transcript_subset.csv”, row.names=FALSE)
write.csv(subset_genes, “fc_gene_subset.csv”, row.names=FALSE)
Thousands of bacterial genome sequences are publicly available. In order to understand the genomic characteristics of these organisms and identify which genes are distinct and common among genomes of interest, it is important to perform a comparative analysis. However, the analysis is often tedious and challenging. This tutorial seeks to address this issue by providing a step-by-step tutorial on how to perform comparative analysis of bacterial genomes on the Zuputo cluster.
The first step is to prepare the analysis environment. We do this by loading Anaconda .
Module load anaconda/3
Configure software channels
conda config --add channels conda-forge conda config --add channels bioconda conda config --add channels daler conda config --add channels defaults
Download the Analysis pipeline
git clone https://github.com/vappiah/bacterial-genomics-tutorial.git
Change directory to the downloaded folder
cd bacterial-genomics
Create conda environment. Packages are listed in the environment.yaml file
conda env create -f environment.yaml
Download the polishing tool pilon
mkdir apps wget https://github.com/broadinstitute/pilon/releases/download/v1.23/pilon-1.23.jar -O apps/pilon.jar
Activate environment
source activate bacterial-genomics-tutorial
Give execution rights for all scripts
chmod +x *.{py,sh,pl}
TIME FOR ANALYSIS
Step 1: Download data
./download_data.sh
Step2: Perform de novo assembly using spades
./assemble.sh
Step3: Polish the draft assembly using pilon. This is meant to improve the draft assembly. The scaffolds will be used. You can also modify the script to use the contigs and compare the result
./polish.sh
Step4: Perform QC for both raw assembly and polished assembly
./qc_assembly.sh

QUAST report showing the quality of draft assembly
Step 5: Reorder contigs against a reference genome using ragtag to generate a draft genome
./reorder_contigs.sh
Step 6: Perform a multi locus sequence typing using MLST software
./mlst.sh
 Step 7: Check for antimicrobial resistance genes using abricate
Step 7: Check for antimicrobial resistance genes using abricate
./amr.sh
 Step 8: Annotate the draft genome using prokka
Step 8: Annotate the draft genome using prokka
./annotate.sh
Get some statistics on the annotation. Features such as genes, CDS will be counted and displayed. This python script requires you to specify the folder where annotations were saved . i.e. P7741
python get_annotat_stats.py P7741
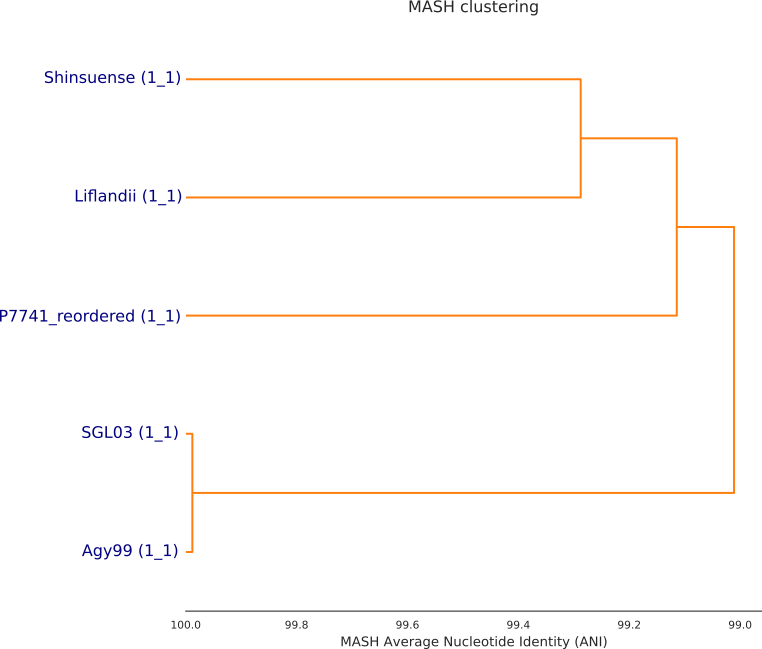
Step 9: Generate dendogram using dREP
./dendogram.sh
Step 10: Perform Pangenome Analysis using Roary. Input files are gff (version 3) format. It is recommended to use prokka generated gff. So we generate the gffs for the files in the genome folder by re-annotating with prokka. We use the get_genome_gffs script
./get_genome_gffs.sh
Then perform pangenome analysis
./get_pangenome.sh
#get gene summary for three of the organism. the default is P7741 Agy99 and SGL03. Feel free to change it. A venn diagram will be generated (gene_summary.jpg)
python get_gene_summary.py P7741 Agy99 and SGL03 pangenome/gene_presence_absence.csv

Step 11: Visualize your genome by generating ring structures using BRIG. This can be done using the video tutorial here

You would want to combine the analysis results into a zip file for download and view locally. After which you can use scp to download to your machine.
zip -r results QC_ASSEMBLY P7741* mlst.csv amr.summary.tab dendogram pangenome *.jpg results
#Now that you have been able to perform a bacterial comparative genome analysis. It's time to apply your skills on real world data.
#Good luck and see you next time
Citation
You just have to declare a statement.
Analysis was done using scripts written by Vincent Appiah, University of Ghana
Gene Expression Analysis Pipeline
Gene expression analysis enables us to identify genes or transcripts that are have significant changes across the conditions. It can often be a daunting task setting up a computational environment to perform this task. This tutorial attempts to reduce this burden by providing a number of already made scripts to perform a gene expression analysis. This document is based on a tutorial by Dave Tang. In this tutorial we make things a bit easier by automating a number of tasks and provide materials which can be used on other datasets by entering a few information. A general workflow of gene expression analysis is shown in the figure below. However, a detailed description of the stages are not provided because that is beyond the scope of this document.

How does the pipeline work?
The entire process begins with creating of config file template, entering of parameters in the config file and executing the pipeline.
To begin the analysis, you first need to load the pipeline via the environmental module.
module load bioinfo/pipelines/dge_1
Then create the config file template with the command below
run_dge create-config
The next stage is to enter your parameters in the config file. Read more in the Parameters section
Afer parameters have been set, you proceed with the execution of the pipeline using the command below
run_dge config.config
Parameters
Parameters are set in the config file. What we did earlier was to prepare a template to in which we will enter the parameters. 12 parameters will have be set (One of them has a default value “None”). What you have to do is to enter the right information at the right side of the equal sign.
Lets take a look at the parameters.
outputdir : this is the directory where you want your results to be saved
ref_gtf : this is your gtf file name. GTF is the annotated reference genome and it is needed for creating the splice sites. You have to specify the fullpath and the file as well. Eg. If your gtf files has the name “mygtf.gtf” and it is located in the directory “/home/user/myfiles”m then the ref_gtf parameter will be “/home/user/myfiles/mygtf.gtf” . In the config file it will be
ref_gtf= “/home/user/myfiles/mygtf.gtf
ref_fasta: This is the reference file in the fasta format. Its parameter will be set in the same way as
files: directory which contains all your sample files.
seqtype: RNAseq sequencing experiment can either be paired ( both forward and reverse strands are sequenced) or single( only forward strand is sequenced). for paired the parameter should be ‘paired’ or ‘p’ . Single will be ‘single’ or ‘s’
threads: this depends on your computer architecture. On our hpc we recommend a maximum thread of 36
phenodata: this file contains the experiment design. It includes the columns for filenames , conditions, confounders. A sample can be found here
covariate= this is the column of interest (it should be included in the phenodata file)
confounders=factors that could affect the results ( for example in measuring the mortality of a disease, one has to take into account age as it can also lead to the death of an individual. Age is therefore a confounding factor). confounders must also be present in the phenodata file.
split_point= this is used to extract names for identification. Usually file names will be used in the experiment design found in the phenodata file. However, file names will have extensions (eg .fastq, .gz) which are absent in the phenodata. Therefore split point indicates the range of position in filenames that should be used. Consider the filename ERR215671_r1.fastq in the experimental design we have ERR215671. in the filename the name we will interested in is ERR215671 which is from position 1 to 9. Therefore our split_point value will be 9.
ext= this is used to select sample files. If your filename is ERR2157671.fastq, then the extension is fastq. The condition for the extension is that it must be the last group of characters in the filename after the dot(.)
mode= used to perform a stagewise analysis. In this pipeline we classify the entire activity into 3 stages: indexing, mapping, differential expression. However the modes are four I.e. i,m,d and a
i – indexing
m – mapping
d – differential expression
a – all ( i.e. i,m, and d)
Demonstration
Step 1: Load the pipeline
module load bioinfo/pipelines/dge_1
Step 2: Create blank config file
run_dge create-config
Step 3:
Enter your parameters in the config file
Step 4: Run analysis
run_dge “config file”
If config file is with the name config.config and in the directory /home/user/files
then the command becomes
run_dge /home/user/files/config.config
Step 5: Check your output directory and find the file dge_results.csv
The file can be downloaded for downstream analysis ( more on this later)
Demonstration with sample data
There is a command to download a sample data for analysis. The command will download files to a directory with the name data which will be found in your current working directory. In addition, it also prepares the config file for you. Use the command below
module load bioinfo/pipelines/dge_1
run_dge download-sample-data
You can then execute the analysis pipeline.
run_dge ./data/config.config
Results will be saved as dge_results.csv in data directory
What tools are used in this pipeline?
HISAT2 v2.1 – indexing and mapping
samtools v1.9 – for sorting indexed files
stringtie v2.0.6– Transcriptome assembly and quantification
gffcompare – compare the StringTie transcripts to known transcripts using
Ballgown (R package) – Differential expression
What are the limitations of this pipeline?
Pipeline was designed to work on only linux environment. The main reason is that most of the tools only work on unix platforms ( samtools, hisat2 and stringtie )
This pipeline was tested with a two–condition experiment and therefore we assume that is what it can do at the moment. In future we will include a feature to handle experiments with three or more conditions.
This pipeline was tested with a single timepoint dataset. In the future we will include a future to handle a time course analysis
Synopsis: Pangenome is the entire set of genes present in all strains within a clade. In this tutorial clade refers to the all genomes on which this analysis is being performed on). The pangenome of a species comprise of the core genome containing genes present in all strains within the clade, the accessory / dispensable genes and strain-specific genes.
In this tutorial we will perform a pangenome analysis on raw bacterial genome specifically Mycobacterium ulcerans P7741 strain together with 4 other trains with known sequence data. This tutorial should be done in a linux environment.
PREPARE SOFTWARE TOOLS
We will use a number of tools to perform the analysis. We will use the conda environment to download and install the tools. Procedure for installing tools is documented here
Create and activate your conda environment. We are using the environment bioinformatics
source activate bioinformatics
Its now time to dig in
STEP 1: DOWNLOAD THE DATA
Our dis hosted on the SRA database. Since we already know the download link we use wget to get it.
wget https://sra-pub-src-1.s3.amazonaws.com/ERR3336325/all_minion.fastq.1
Rename the sample to ERR3336325
mv all_minion.fastq.1 ERR3336325.untrimmed.fastq
STEP 1: TRIM ADAPTERS
We will first trim adapters for the oxford nanopore data using porechop tool (Install with anaconda)
We will use 18 threads
threads=18
porechop -i ERR3336325.untrimmed.fastq -o ERR3336325.fastq --format fastq -t $threads
STEP 2: DE NOVO ASSEMBLY
We then perform a de novo assembly on our sample ERR3336325. The sample we are using was sequenced using an oxford nanopore MinION device so we will use the tool called flye to do that.
Activate your conda environment and install flye (view the procedure here)
source activate bioinformatics
Before we start the de novo assembly lets create our output directory
flye
mkdir output
Now execute flye the 10 threads
flye --nano-raw ERR3336325.fastq -o output -g 5.6m -t 10 -i 2
Once the de novo assembly is done the contigs will be located in output as assembly.fasta
Lets check.
ls output
STEP 3: ANNOTATION
After de novo assembly we will annotate the contig using the tool prokka
prokka --cpus 10 output/assembly.fasta --prefix ERR3336325
When this is completed, you will have folder called ERR3336325 created. In this folder you will have 12 files created :
ERR3336325.err, ERR3336325.faa, ERR3336325.ffn, ERR3336325.fna, ERR3336325.fsa, ERR3336325.gbk, ERR3336325.gff, ERR3336325.log, ERR3336325.sqn, ERR3336325.tbl, ERR3336325.tsv, ERR3336325.txt
Check with the ls command
ls ERR3336325
The files include sequence data for nucleotide (.fna) and amino acids(.faa) as well as annotated files (.gbk and .gff)
We can look up the results to get some interesting information such as number of genes identified as well as counts of proteins, RNAs,etc
Lets get how many annotations was done in the genome
grep '>' ERR3336325/ERR3336325.ffn|wc -l 7002
Lets get the number of proteincoding genes
grep '>' ERR3336325/ERR3336325.faa |wc -l 6929
Lets get the number of hypothetical proteins the genome
grep 'hypothetical protein' ERR3336325/ERR3336325.faa |wc -l 4497
Find the number of operons
grep -i 'operon' ERR3336325/ERR3336325.ffn |wc -l 0
STEP 4: PANGENOME ANALYSIS
Pangenome analysis will be done using the tool roary
This analysis will make use of gff files.
At the moment we only have gff file for our sample ERR3336325. This is located in the prokka results directory as ERR3336325.gff.
Lets make directory to keep the gffs that will be used for the analysis.
mkdir gffs
Copy the ERR3336325 sample
cp ERR3336325/ERR3336325.gff gffs ls gffs
We would like to perform this analysis together with other strains of Mycobacterium ulcerans (i.e. Agy99, S4018, CSURQ0185 and Harvey
Note that roary uses gff files generated by prokka for the pangenome analysis. So we have to prepare gff files for the 4 other strains we will be using. This will involve first downloading the fasta files and preparing the gff files using prokka
Lets download the fasta files for the 4 strains wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/013/925/GCA_000013925.2_ASM1392v2/GCA_000013925.2_ASM1392v2_genomic.fna.gz wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/001/870/585/GCA_001870585.1_ASM187058v1/GCA_001870585.1_ASM187058v1_genomic.fna.gz wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/524/035/GCA_000524035.1_ASM52403v1/GCA_000524035.1_ASM52403v1_genomic.fna.gz wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/902/506/705/GCA_902506705.1_Q0185/GCA_902506705.1_Q0185_genomic.fna.gz wget ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/026/445/GCA_000026445.2_ASM2644v2/GCA_000026445.2_ASM2644v2_genomic.fna.gz
Lets uncompress the files
gunzip GCA_000013925.2_ASM1392v2_genomic.fna.gz gunzip GCA_001870585.1_ASM187058v1_genomic.fna.gz gunzip GCA_000524035.1_ASM52403v1_genomic.fna.gz gunzip GCA_902506705.1_Q0185_genomic.fna.gz gunzip GCA_000026445.2_ASM2644v2_genomic.fna.gz
Lets prepare the gff files using prokka
prokka --cpus 10 --kingdom Bacteria --outdir Agy99 --genus Mycobacterium GCA_000013925.2_ASM1392v2_genomic.fna prokka --cpus 10 --kingdom Bacteria --outdir S4018 --genus Mycobacterium GCA_001870585.1_ASM187058v1_genomic.fna prokka --cpus 10 --kingdom Bacteria --outdir Harvey --genus Mycobacterium GCA_000524035.1_ASM52403v1_genomic.fna prokka --cpus 10 --kingdom Bacteria --outdir CSURQ0185 --genus Mycobacterium GCA_902506705.1_Q0185_genomic.fna prokka --cpus 10 --kingdom Bacteria --outdir Liflandii --genus Mycobacterium GCA_000026445.2_ASM2644v2_genomic.fna
The gff files will be located in the individual output directory
Verify with ls command.
ls Agy99 ls S4018 ls Harvey ls CSURQ0185 ls Liflandii
Now, lets copy those gff files to our gffs directory
cp Agy99/PROKKA*.gff gffs/Agy99.gff cp S4018/PROKKA*.gff gffs/S4018.gff cp Harvey/PROKKA*.gff gffs/Harvey.gff cp CSURQ0185/PROKKA*.gff gffs/CSURQ0185.gff cp Liflandii/PROKKA*.gff gffs/Liflandii.gff
Lets verify that all files are present in the gffs directory
ls gffs
Its time for the main course meal. I assume that you already have roary installed and setup
We generate a pangenome with a core gene alignment using 10 threads
roary -f roaryresult -p 10 -e -n -v --mafft gffs/*.gff
Visualizing the results
This involves two stages
– generating trees ( in newick format) using FastTree
– plot the trees using python script
FastTree -nt -gtr roaryresult/core_gene_alignment.aln>roaryresult/mytree.newick
./roary_plots.py --labels roaryresult/mytree.newick roaryresult/gene_presence_absence.csv mv *.png roaryresult
Once this is complete three png files will be created.
ls roaryresult
View those plots to understand your data. Other useful outputs will be located in the roaryresult directory.
Synopsis: This document demonstrates each step of the GATK pipeline that can be used to call out variants in whole exome data. It follows the GATK best practices . We assume that you are familiar with the linux environment
We first load our tools
module load bioinfo/samtools/1.9 module load bioinfo/gatk/4.1 module load bioinfo/bwa/0.7 module load java/8 module load R/3.4.3 module load bioinfo/tabix/1.10.2 module load bioinfo/bgzip/1.10.2 picard=/opt/apps/bioinfo/picard/2.23.1/picard.jar Let's download the reference and annotation files. We will use hg19bundle from broadinstitute
mkdir hg19bundle
wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/1000G_omni*wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/hapmap_3.3*wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/Mills_and_1000G*wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/1000G_phase1.snps.high_confidence*wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/dbsnp_138* wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/1000G_phase1.indels.hg19.sites.vcf.gz wget --directory-prefix=hg19bundle ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/ucsc.hg19*
We Process the downloaded files.
unzip the reference genome
gunzip hg19bundle/ucsc.hg19.*.gz
Unzip the annotation files( After several experimentations this was the best approach I could come up with)
gunzip hg19bundle/1000G_phase1.snps.high_confidence.hg19.sites.vcf.idx.gz gunzip hg19bundle/1000G_phase1.snps.high_confidence.hg19.sites.vcf.gz gunzip hg19bundle/dbsnp_138.hg19.vcf.idx.gz gunzip hg19bundle/dbsnp_138.hg19.vcf.gz gunzip hg19bundle/hapmap_3.3.hg19.sites.vcf.idx.gz gunzip hg19bundle/hapmap_3.3.hg19.sites.vcf.gz gunzip hg19bundle/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf.idx.gz gunzip hg19bundle/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf.gz gunzip hg19bundle 1000G_omni2.5.hg19.sites.vcf.idx.gz gunzip hg19bundle/1000G_omni2.5.hg19.sites.vcf.gz
Let's set the reference and annotation file names
ref=hg19bundle/ucsc.hg19.fasta
GCONF=hg19bundle/1000G_phase1.snps.high_confidence.hg19.sites.vcf DBSNP=hg19bundle/dbsnp_138.hg19.vcf HAPMAP=hg19bundle/hapmap_3.3.hg19.sites.vcf MILLS=hg19bundle/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf OMNI=hg19bundle/1000G_omni2.5.hg19.sites.vcf
We will use 18 threads for this task
threads=18
Let us download the exome raw sequence reads. The data is taken from the 1000 genomes project repository
mkdir reads
Let's rename the raw reads (We move to the reads directory and rename )
mv SRR100027_1.filt.fastq.gz reads/read1.fastq.gz mv SRR100027_2.filt.fastq.gz reads/read2.fastq.gz
We also download the capture file which contains the regions of the genome that were sequenced
wget --directory-prefix=hg19bundle ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/exome_pull_down_targets/20130108.exome.targets.bed
Set the location
targetregion=$resourcedir/20130108.exome.targets.bed
Let's set location for java temp directory
javatmp=tmp
mkdir $javatmp
Set path to the data input and output files
read1=reads/read1.fastq.gz read2=reads/read2.fastq.gz
outdir=outdir mkdir $outdir
Now that we have all the tools and resources setup. Let's start the variant calling.
Step 1: We begin our pipeline with indexing the reference genome
bwa index $ref
Step 2:
samtools faidx $ref
Note: GATK provides index files and so if you have them, you can skip step 1 and 2
For this tutorial you can skip step2
Step 3: Align the fastq files to the reference genome. We use hg19 as teh reference genome
Aligning also requires that we add readgroups . So let's define our readgroups
PLATFORM="illumina" sampleid=S01
(If you are running several samples , then you should generate unique ids for each sample)
samplename=NGR
bwa mem -t $threads -R "@RG\\tID:${sampleid}\\tPL:$PLATFORM\\tPU:None\\tLB:None\\tSM:${samplename}" $ref $read1 $read2 > $outdir/reads.sam
We convert the samfile to bamfile
samtools view -h $outdir/reads.sam -O BAM -o $outdir/reads.bam
Sort the bamfile
samtools sort -O BAM --reference $ref -@ $threads -o $outdir/reads.sorted.bam $outdir/reads.bam
Index the bamfile
samtools index $outdir/reads.sorted.bam
Step 4. Variant Calling with GATK.
#We first mark duplicates. This step is computationally expensive so we try to speed it using gatk-spark #module
java -jar $picard MarkDuplicates \ I=$outdir/reads.sorted.bam \ O=$outdir/mark_dup.bam \ M=$outdir/dum_metrics.txt
gatk BaseRecalibrator -I $outdir/mark_dup.bam -R $ref --known-sites "$GCONF" --known-sites "$DBSNP" --known-sites "$MILLS" -O $outdir/reads.recal.table gatk ApplyBQSR -R $ref -I $outdir/mark_dup.bam -O $outdir/reads.recal.bam -bqsr $outdir/reads.recal.table
#variant calling
gatk --java-options "-Xmx4g" HaplotypeCaller -R $ref -I $outdir/reads.recal.bam -L $targetregion --dbsnp $DBSNP --native-pair-hmm-threads $threads --lenient true -O $outdir/reads.g.vcf
- Kwofie, S.K.; Enninful, K.S.; Yussif, J.A.; Asante, L.A.; Adjei, M.; Kan-Dapaah, K.; Tiburu, E.K.; Mensah, W.A.; Miller, W.A., III; Mosi, L.; Wilson, M.D. (2019). Molecular Informatics Studies of the Iron-Dependent Regulator (ideR) Reveal Potential Novel Anti-Mycobacterium ulcerans Natural Product-Derived Compounds. Molecules, 24, 2299.
- Kwofie, S. K.; Broni, E.; Teye, J.; Quansah, E.; Issah, I.; Wilson, M. D.; Miller, W. A.; Tiburu, E. K; Bonney, J. H. K. (2019). Pharmacoinformatics-Based Identification of Potential Bioactive Compounds against Ebola Virus Protein VP24. Computers in Biology and Medicine, 2019, 103414. https://doi.org/10.1016/j.compbiomed.2019.103414.
- Kwofie, S.K.; Dankwa, B.; Enninful, K.S.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M.D. (2019). Molecular Docking and Dynamics Simulation Studies Predict Munc18b as a Target of Mycolactone: A Plausible Mechanism for Granule Exocytosis Impairment in Buruli Ulcer Pathogenesis. Toxins, 11, 181.
- Kwofie, S. K., Dankwa, B., Odame, E. A., Agamah, F. E., Doe, L. P. A., Teye, J., Agyapong, O., Miller, W.A., III, Mosi, L., Wilson, M.D. (2018). In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules (Basel, Switzerland), 23(7)
- Morang’a CM, Amenga-Etego L, Bah SY, Appiah V, Amuzu DS, Amoako N, Abugri J, Oduro AR, Cunnington AJ, Awandare GA, Otto TD (2020). Machine learning approaches classify clinical malaria outcomes based on haematological parameters. medRxiv 2020.09.23.20200220; https://doi.org/10.1101/2020.09.23.20200220



